


PIPELINE MODE USAGE
3.1.- CONFIGURING AND RUNNING PIPELINES
The pipeline mode of “VariantSeq” is a pipeline configuration system that let the user to execute all the steps of a pipeline, automatically one after the other (Fig.44). To perform a Variant-Seq analysis using the pipeline mode, the user must access the VariantSeq protocols tab of the Top menu and then select the option Pipeline Mode. Then a first pipeline interface will appear summarizing all currently available pipelines based on Command Line Interface (CLI) software offered by the Pipeline mode of “VariantSeq”.
To perform an analysis using the pipeline mode, please go to [ Variant calling Protocols → Pipeline Mode ] and follow Fig. 44
The experiment configuration panel has two tabs: “Choose pipeline” and “Input and Configuration”. “Choose pipeline” enables users to create a workflow project and select the pipeline from a list. “Input and Configuration” enables the user to upload input files, adjust options and parameters, and set an output folder. The procedure is shown in Fig. 44.
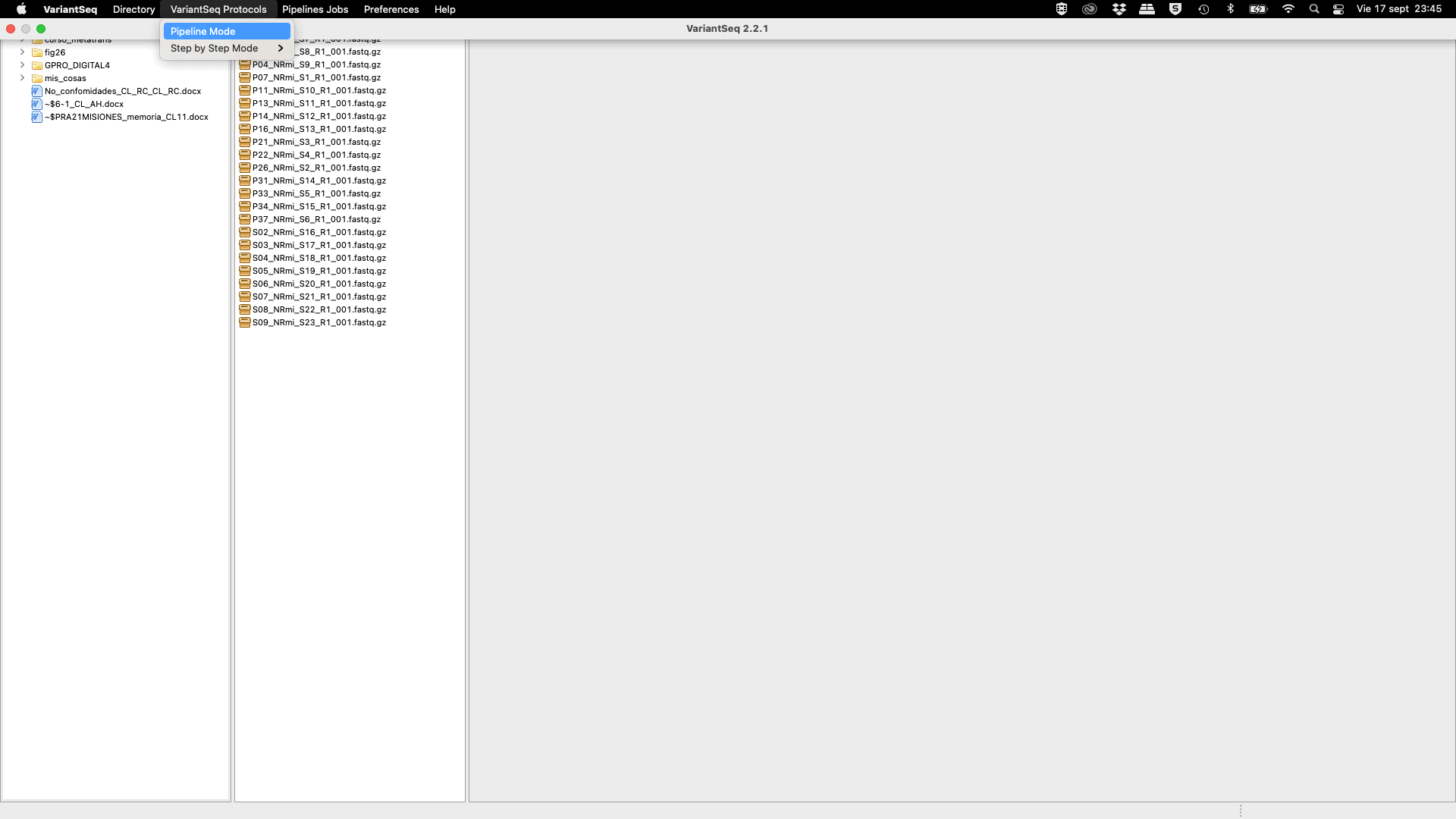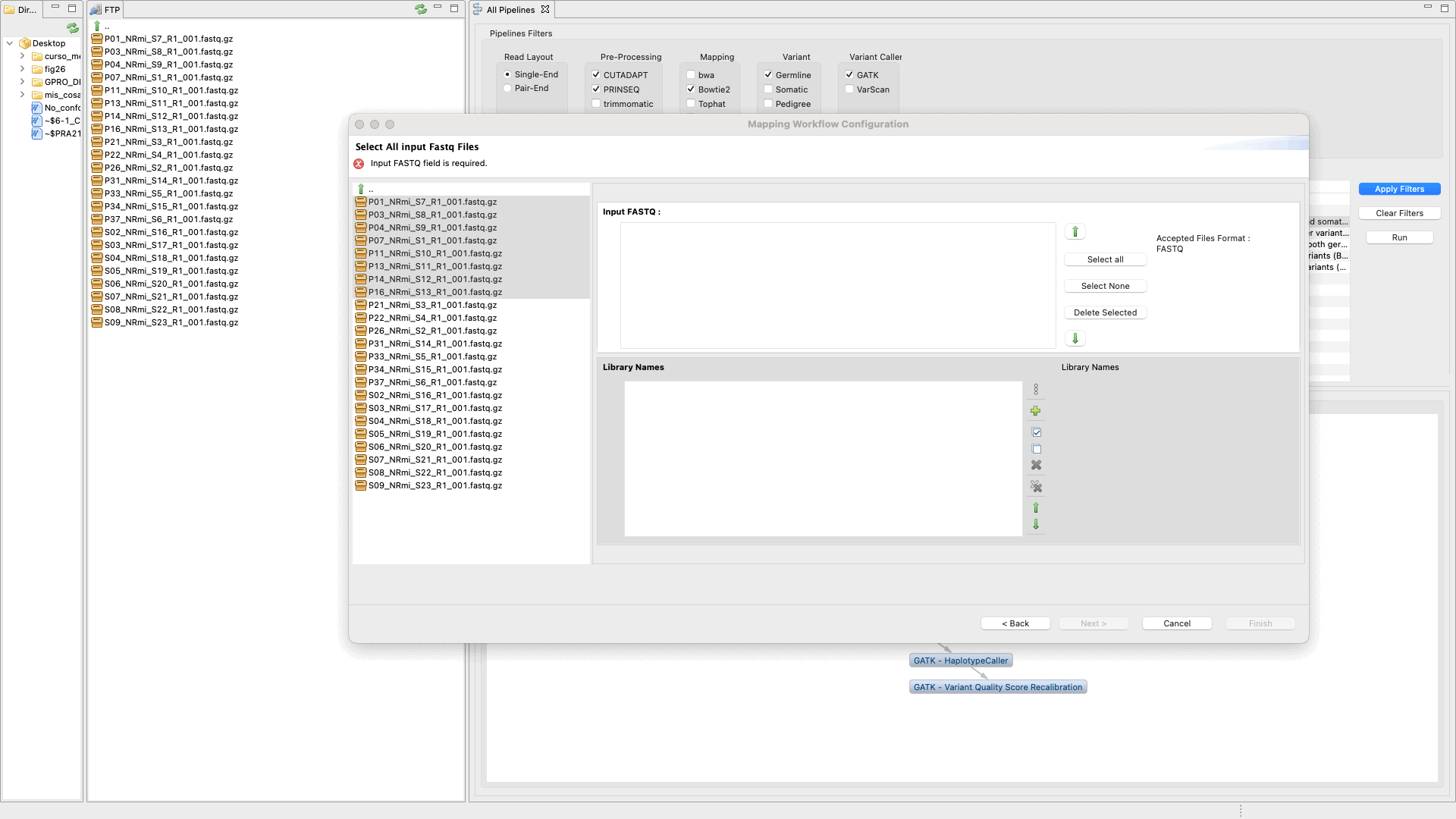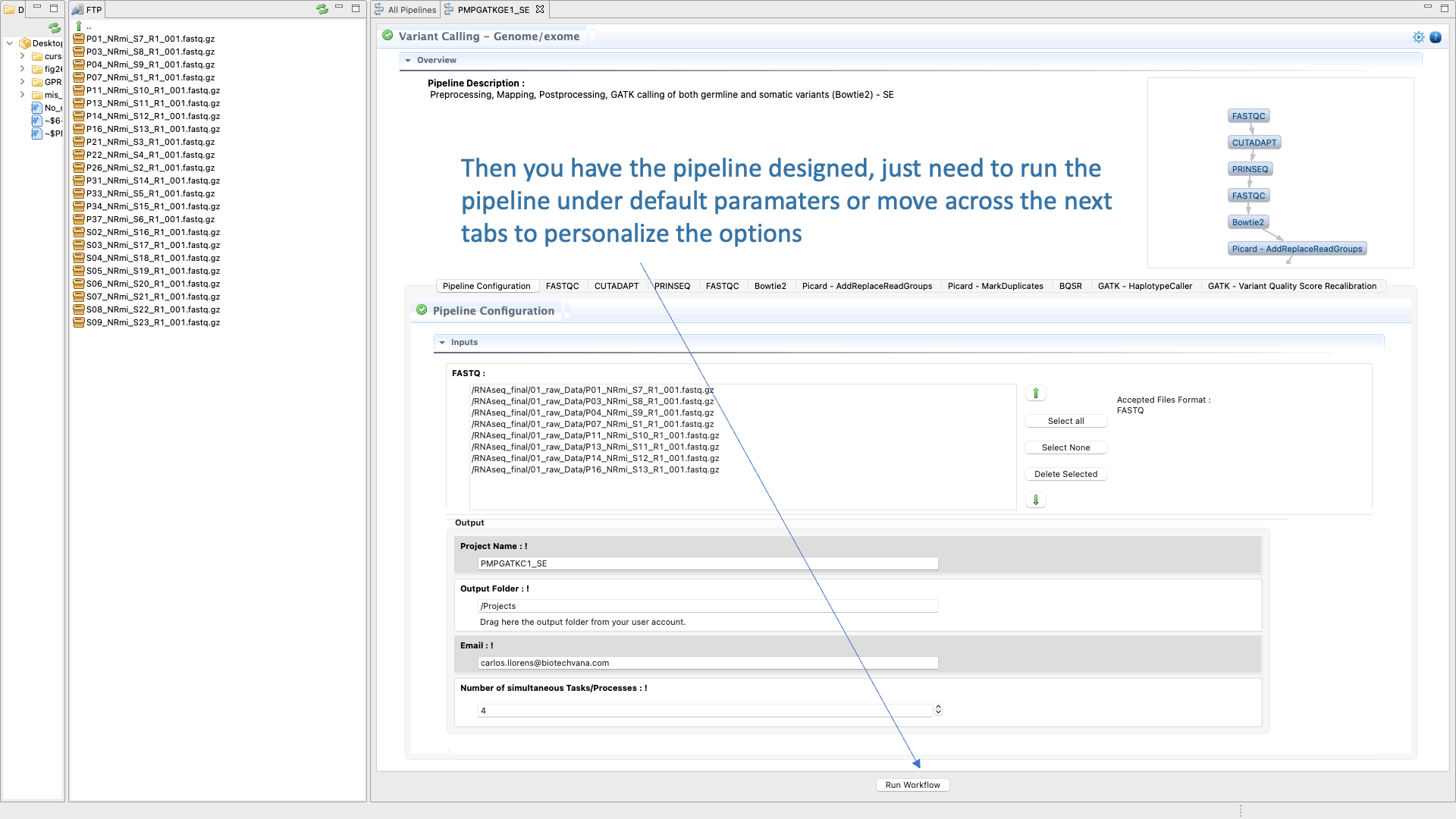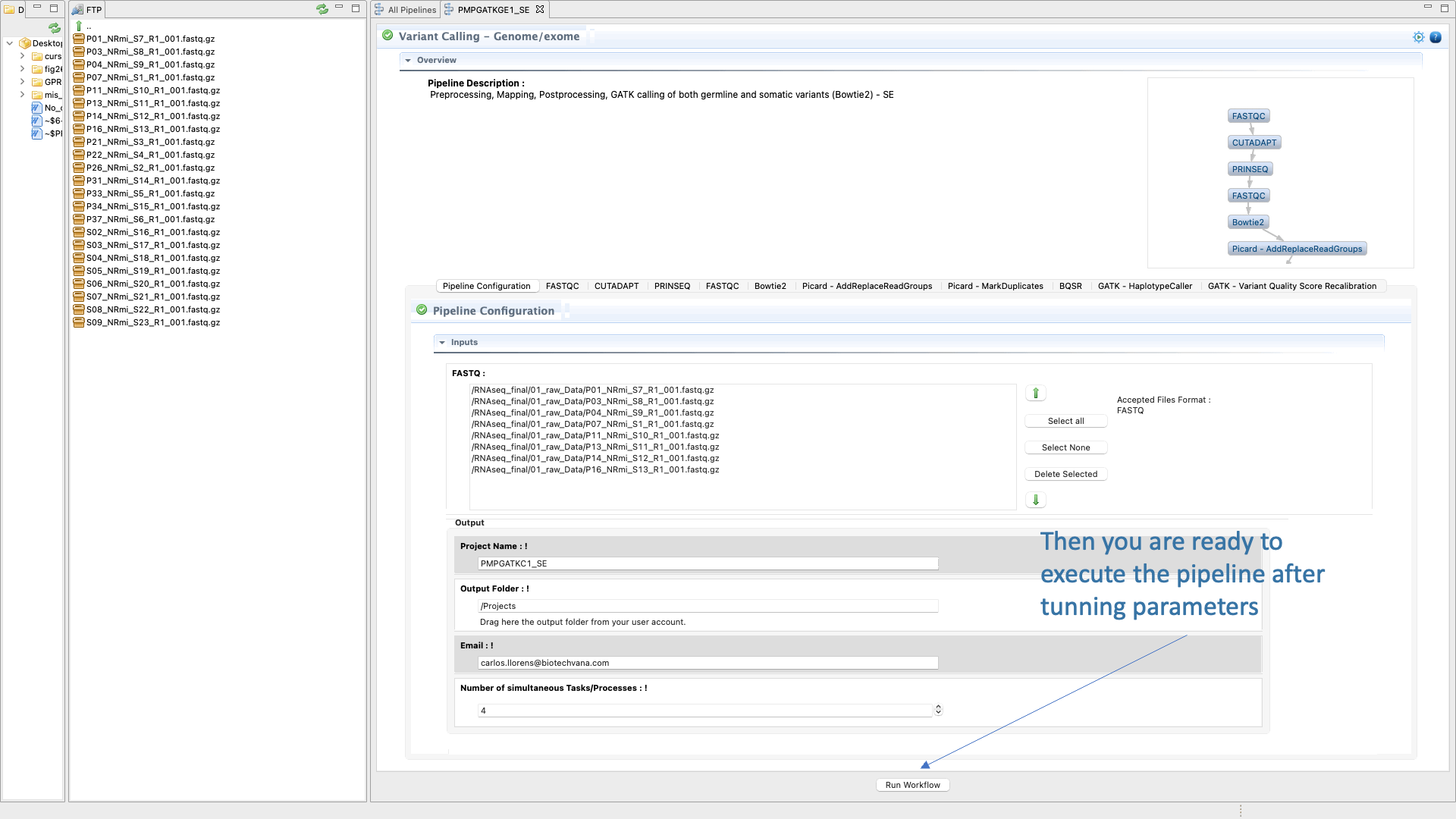
Figure 44: Example of configuration and execution for a Variant-seq analysis using the pipeline mode.
Using the pipeline mode you only need to select one pipeline from the list and then configure the experiment going to a set of nested interfaces to;
- upload any input data or material needed by the pipeline (i.e. fastq files, reference genome or training sets, etc)
- declare the outputs
- declare the experiment design design (samples, groups, etc)
- configure the parameters and options of each CLI tool used in each step (for example, “FastQC” for quality analysis, “Trimmomatic” for preprocessing, “BWA” for mapping, “GATK” for base quality score recalibration, “Picard Tools” for marking duplicates and “GATK” for for haplotype calling).
- finally run the pipeline .